Research areas
We work on computational materials science at the intersection of artificial intelligence and atomistic simulation, with an emphasis on the predictive design of functional materials. We develop AI-accelerated frameworks that enable efficient and systematic exploration of high-dimensional materials design spaces. Our methodology combines deep reinforcement learning, machine-learned interatomic potentials, and automated simulation workflows to advance atomistic modeling and simulation for materials discovery. We seek to understand and predict materials formation, structural evolution, and functional behavior, while addressing fundamental questions related to chemical bonding, materials processing, and ion and defect transport. We apply these computational approaches to a broad range of materials systems, including electronic materials for heterojunctions, ionic conductors for energy storage applications, and heterogeneous (electro)catalysts.
인공지능(AI)과 원자론적 시뮬레이션(Atomistic Simulation)의 접점에서 계산 재료 과학을 연구하며, 특히 기능성 재료의 예측 설계에 중점을 두고 있습니다. 우리는 고차원 재료 설계 공간을 효율적이고 체계적으로 탐색할 수 있는 AI 가속 프레임워크를 개발합니다. 우리는 강화 학습, 머신러닝포텐셜, 그리고 자동화된 시뮬레이션 워크플로우를 결합하여, 재료 발견을 위한 원자론적 모델링 및 시뮬레이션 기술을 선도하고 있습니다. 또한 재료의 형성 과정, 구조적 진화, 기능적 거동을 이해하고 예측하는 것을 목표로 하며, 화학 결합, 재료 공정, 이온 및 결함 수송과 관련된 근본적인 문제들을 해결하고자 합니다.
Agentic computational workflows

Leveraging large language models (LLMs), we develop scientific AI agents for automated computational workflows. Our goal is to facilitate materials screening and discovery by integrating databases, machine learning models, and high-throughput pipelines. These agentic workflows are applied to problems in energy, sustainability, and electronic materials.
대규모 언어 모델을 활용하여 자동화된 계산 워크플로우를 위한 과학용 AI 에이전트를 개발합니다. 데이터베이스, 머신러닝 모델, 그리고 고성능 파이프라인을 통합하여 소재 스크리닝 및 발견을 촉진하고자 하며, 이러한 에이전트 기반 워크플로우는 에너지, 지속 가능성, 그리고 전자 재료 분야의 문제 해결에 적용됩니다.
Agentic atomistic simulation

We develop agentic atomistic simulation frameworks that overcome the timescale and sampling limitations of conventional atomistic methods. By coupling machine-learned interatomic potentials (MLIPs) with reinforcement learning, we enable efficient exploration of atomic transition pathways. These approaches are applied to diverse materials phenomena, including ion transport in ionic conductors, phase transformations, and materials processing processes such as thermal annealing.
기존 원자 수준 모델링 방법론의 시간적, 샘플링 한계를 극복하는 에이전트 기반 원자 수준 시뮬레이션 프레임워크를 개발합니다. 머신러닝 포텔셜과 강화학습을 결합하여 원자 전이 경로를 효율적으로 탐색할 수 있도록 하며, 이러한 접근법은 이온 전도체의 이온 수송, 상전이, 열처리 등 다양한 재료 현상에 적용됩니다.
Modelling and simulations
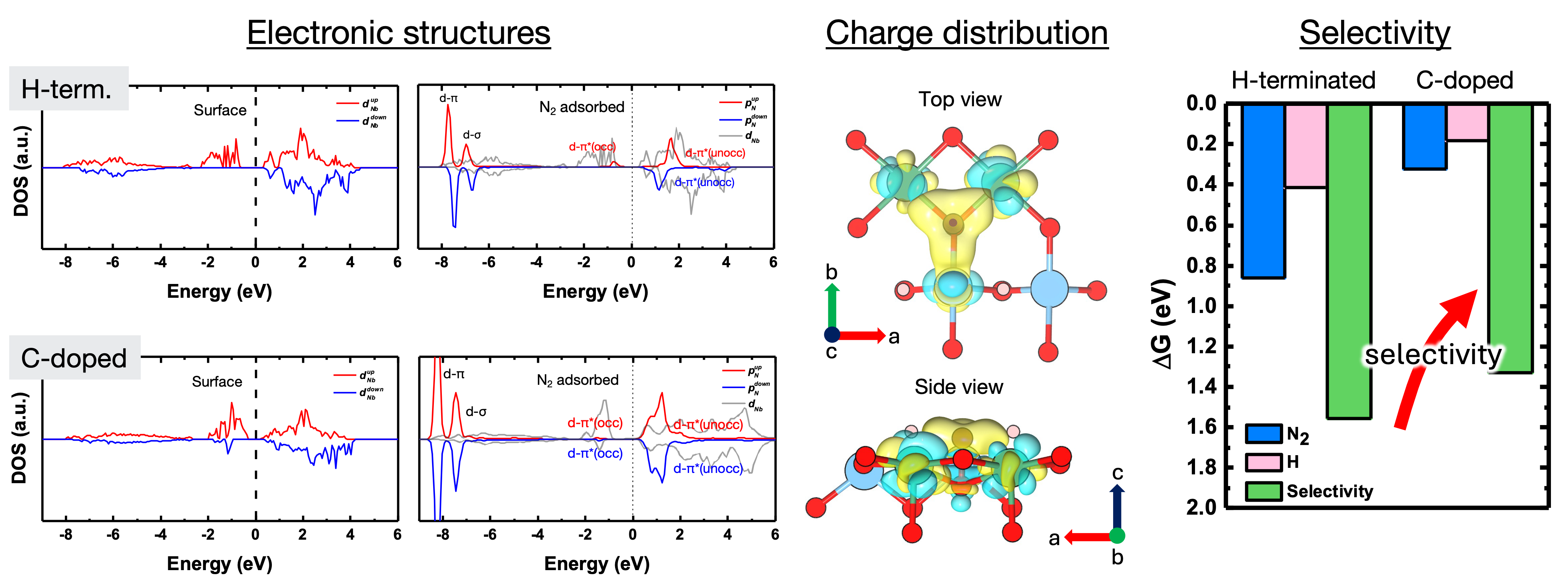
We employ first-principles (ab initio) calculations to uncover atomic-scale mechanisms governing materials behavior. We model materials at various scales and combine electronic structure theory, chemical bonding analysis, and ab initio thermodynamics to elucidate reaction mechanisms and establish quantitative structure–property relationships. Furthermore, we utilize ab initio molecular dynamics to analyze the dynamic behavior of materials.
원자 수준에서 재료의 거동을 지배하는 메커니즘을 규명하기 위해 제일원리 계산을 수행합니다. 재료를 다양한 스케일에서 모델링하고 전자 구조 이론, 화학 결합 분석, 그리고 제일원리 열역학을 통합하여 반응 메커니즘을 명확히 하고 정량적인 구조-물성 상관관계를 이해합니다. 또한 제일원리 분자동역학을 통해 재료의 동적 거동을 분석합니다.